Mass Spec: Adducts, Multiply charged species, isotope patterns
Fragments, Adducts, charge
When creating/editing a substance that’s used for Mass Spec you can set
which ions are used by Peaksel: 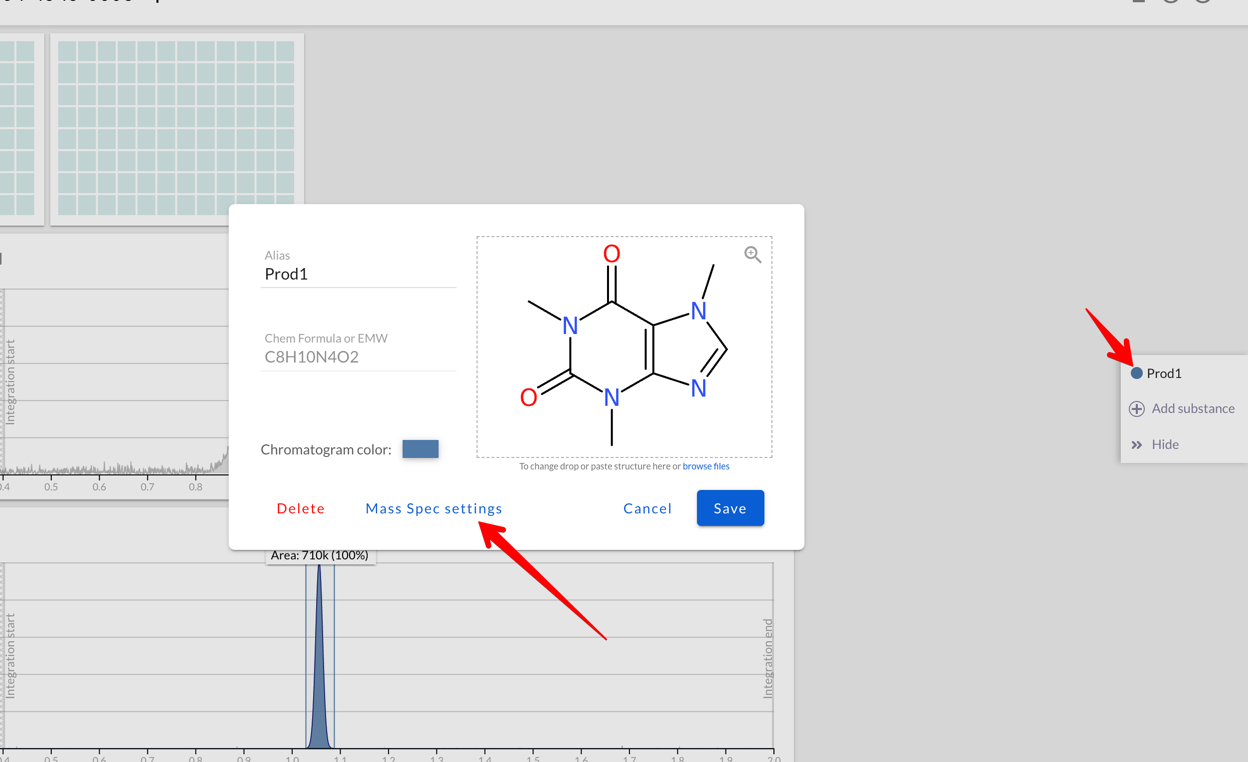
In the Mass Spec settings you can:
-
Add/subtract fragments to/from your initial structure
-
Set max charge count to define how many times you expect your compound to be ionized, see Multiply-charged species for more details.
When extracting a chromatogram, now Peaksel will add up all the
intensities of all the adducts: 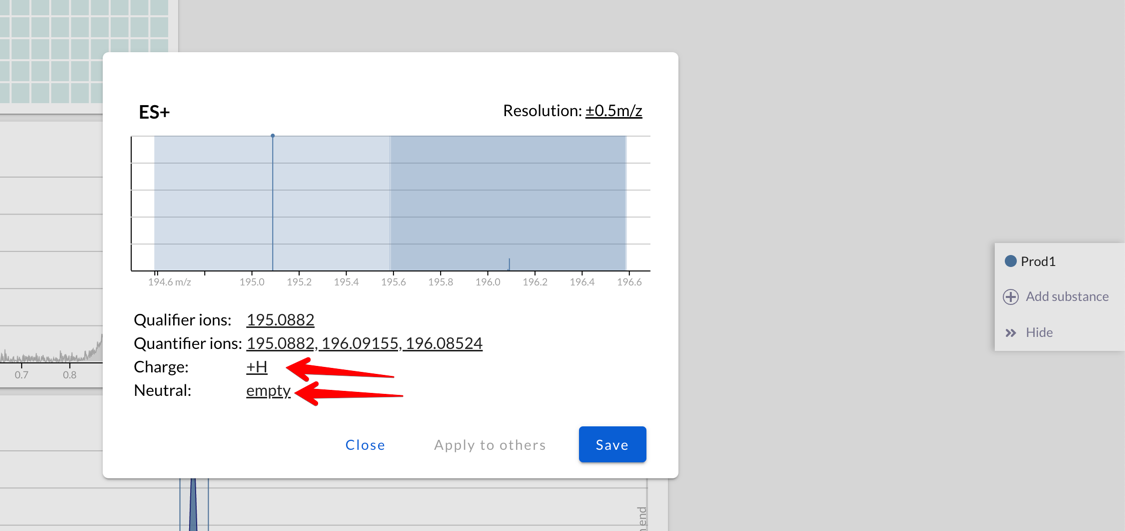
It’s possible to set multiple fragments to be added to or removed from
the compound. They can be charged (like H+, Na+, Cl- ions)
or neutral (like deprotected compound): 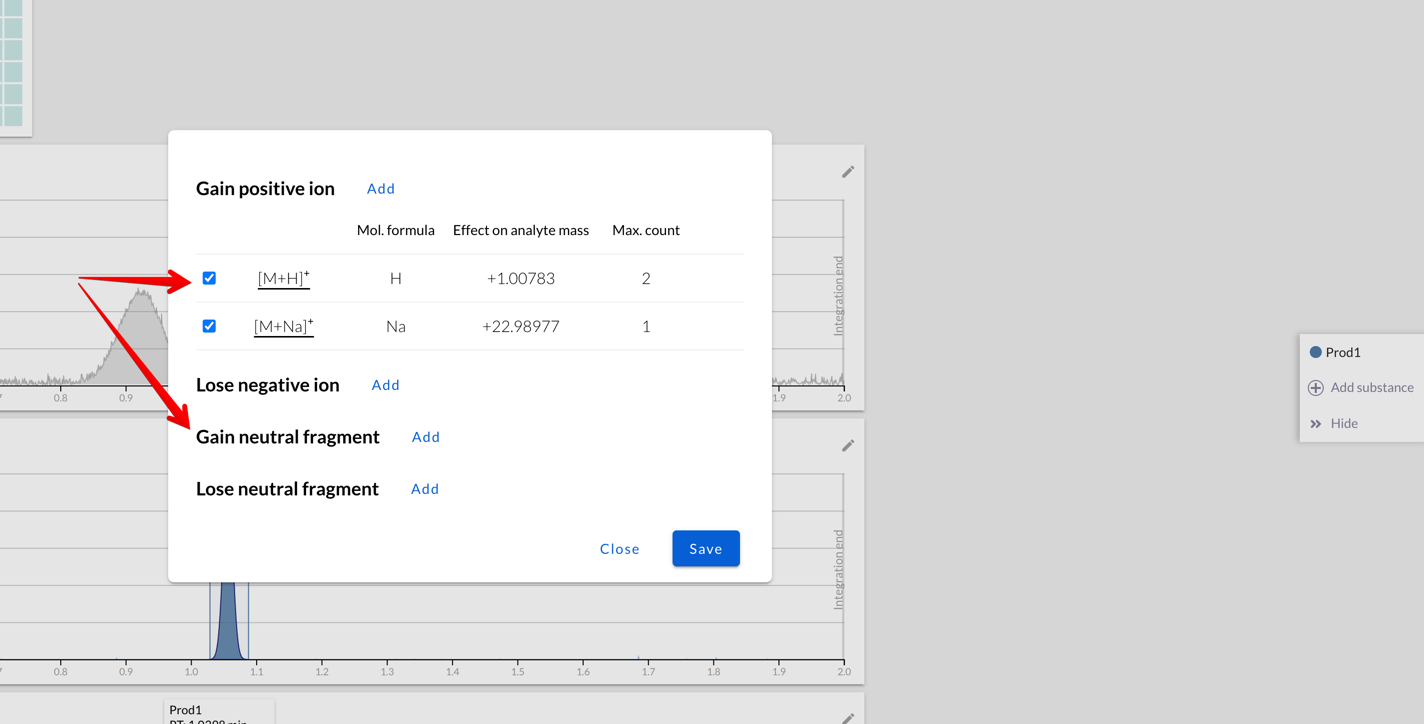
Isotopic patterns, qualifier and quantifier ions
It’s possible to fine-tune which of the ions that Peaksel calculated
should be used: 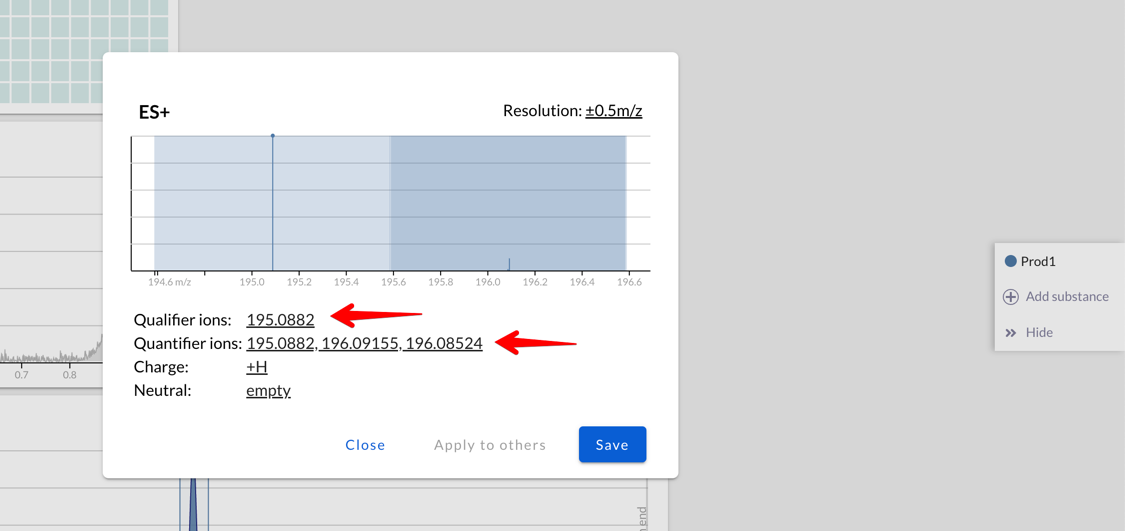
Some use cases:
-
You may want to use one set of ions as quantifiers (they’ll be part of the peak area), while others are used as qualifiers to determine if the peak is actually our compound and not some isobar with very similar mass.
-
Or maybe you work with isopure compounds, and you want to uncheck all but one isotope from quantification
By default, Peaksel will use only the most abundant isotopes (of the
same adduct!) as qualifiers, but only if their predicted intensities are
higher than the tolerance level: 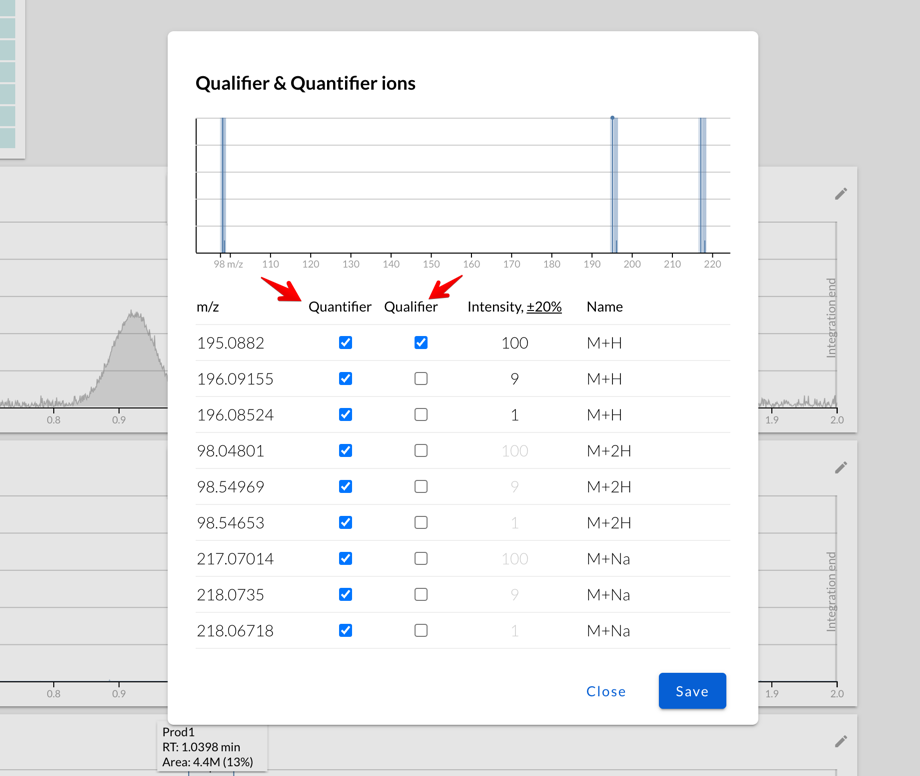
Isotopic matching algorithm
First, Peaksel generates a list of possible ions and their relative intensities using
isotope-distribution library. This is done for all possible
adducts registered, including neutral gains/losses and various charge counts. However, it’s impossible to compute
the relative intensities between 2 adducts, so you (the user) need to determine which of the adduct is going to be
used for the matching. Peaksel defaults to [M+H]+, singly charged.
Additionally, we need a peak spectrum. So first, a chromatographic peak is detected on the EIC, then Peaksel goes through each point of the peak and calculates its mean spectrum.
Now that we know the theoretical ratios of isotopes and the peak spectrum, we can start the actual matching:
-
First, we extract the measured intensities out of the peak spectrum. For this we search for m/z values of interest, however they never match the theoretical m/z exactly. So we search for them within some ± Mass Resolution defined in the Mass Settings dialog.
-
It’s possible there are multiple measurements within the
m/z ± range, and it’s not clear which value must be used. Peaksel chooses the highest intensity for this. However, if the mass gets shifted between 2 measurements of the mass spec (which is a possibility), the intensity ratio won’t necessarily match the theoretical pattern even though it’s the right analyte - this is one of the limitations of the current approach. -
Now we normalize the extracted intensities so that the highest one equals 1.
-
The last step is to go through every m/z from the theoretical and expected intensities and compare them. Only the m/z values that are marked as Qualifiers will be considered. And the spectrum will be considered matching if every Qualifier’s theoretical intensity matches the measured one within some boundaries - the Ratio threshold that’s configured in the Ions Dialog.
Mass Spec resolution
Mass resolution in Peaksel is defined as simple range ±[units of m/z]. So if the analyte’s expected m/z is 101.3 with a ±.5 window, then any signal within 100.8-101.8 is considered to belong to that analyte during mass spec processing.
In MS1:
-
All the Quantifier ions within that range are summed up to form the area of the chromatogram
-
As for the Qualifiers, the highest value in the range is considered when doing isotope matching
In Product Ion Scan:
-
During chromatogram extraction, each scan’s precursor is matched with the expected analyte m/z. If the precursor is within the expected range, then this point and its spectra are used for the extraction.